vlisi · GitHub
Last updated:2026-03-13 12:43
Alluvial Comparison
This script shows how to produce a version of the alluvial comparison panel (MARLIN vs DX vs ALMA), matching the classification-flow view from the full analysis workflow. This code reproduces the base graphic reported in the article Single-workflow Nanopore whole genome sequencing with adaptive sampling for accelerated and comprehensive pediatric cancer profiling.
Overview
The plot compares three aligned classification stages:
MARLINpredicted classDXmolecular diagnosis (reference)ALMApredicted class
Flow width represents sample counts moving through the three stages. This post renders four figures in sequence: combined, ALL-only, AML-only, and a final stacked panel.
Libraries
library(dplyr)
##
## Attaching package: 'dplyr'
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
library(ggplot2)
library(ggalluvial)
library(patchwork)
Define variables
`%nin%` <- Negate(`%in%`)
lineage_from_label <- function(x) {
ifelse(
grepl("^B-ALL", x), "B-ALL",
ifelse(grepl("^T-ALL", x), "T-ALL", "AML")
)
}
pick_alt_within_lineage <- function(current_label, all_levels) {
current_lineage <- lineage_from_label(current_label)
same_lineage_levels <- all_levels[lineage_from_label(all_levels) == current_lineage]
candidate_levels <- same_lineage_levels[same_lineage_levels != current_label]
if (length(candidate_levels) == 0) {
current_label
} else {
sample(candidate_levels, 1)
}
}
lineageCols <- c(
"B-ALL" = "#8caaee",
"T-ALL" = "#ca9ee6",
"AML" = "#ef9f76"
)
metClassColors <- c(
"B-ALL" = "#8caaee",
"T-ALL" = "#ca9ee6",
"AML" = "#ef9f76",
"B-ALL::ETV6-RUNX1" = "#74c7ec",
"B-ALL::DUX4" = "#89dceb",
"T-ALL::TLX1" = "#b4befe",
"AML::NPM1" = "#fab387"
)
Produce simple test data
set.seed(2026)
n_samples <- 180
dx_levels <- c(
"B-ALL",
"T-ALL",
"AML",
"B-ALL::ETV6-RUNX1",
"B-ALL::DUX4",
"T-ALL::TLX1",
"AML::NPM1"
)
dx_weights <- c(
0.18,
0.14,
0.12,
0.20,
0.14,
0.10,
0.12
)
dx_df <- tibble(
sample_id = sprintf(
"S%03d",
seq_len(n_samples)
),
Molecular.Diagnosis = sample(
dx_levels,
n_samples,
replace = TRUE,
prob = dx_weights
)
) %>%
mutate(
Diagnosis = case_when(
grepl("^B-ALL", Molecular.Diagnosis) ~
"B-ALL",
grepl("^T-ALL", Molecular.Diagnosis) ~
"T-ALL",
TRUE ~ "AML"
)
)
# Simulate MARLIN and ALMA predictions from DX labels.
# `runif` thresholds control concordance with DX
# (MARLIN ~78%, ALMA ~72%) and introduce off-label noise.
allLeukData <- dx_df %>%
rowwise() %>%
mutate(
mcf = ifelse(
runif(1) < 0.78,
Molecular.Diagnosis,
pick_alt_within_lineage(
Molecular.Diagnosis,
dx_levels
)
),
ALMA.v0.1.14 = ifelse(
runif(1) < 0.72,
Molecular.Diagnosis,
pick_alt_within_lineage(
Molecular.Diagnosis,
dx_levels
)
),
lineage = case_when(
grepl("^B-ALL", mcf) ~ "B-ALL",
grepl("^T-ALL", mcf) ~ "T-ALL",
TRUE ~ "AML"
),
ALMA.Lineage = case_when(
grepl("^B-ALL", ALMA.v0.1.14) ~
"B-ALL",
grepl("^T-ALL", ALMA.v0.1.14) ~
"T-ALL",
TRUE ~ "AML"
)
) %>%
ungroup() %>%
mutate(
match.Class = ifelse(
mcf == Molecular.Diagnosis,
"TRUE",
"FALSE"
),
ALMA.match.class = ifelse(
ALMA.v0.1.14 == Molecular.Diagnosis,
"TRUE",
"FALSE"
)
)
head(allLeukData)
## # A tibble: 6 × 9
## sample_id Molecular.Diagnosis Diagnosis mcf ALMA.v0.1.14 lineage
## <chr> <chr> <chr> <chr> <chr> <chr>
## 1 S001 AML AML AML AML AML
## 2 S002 B-ALL::DUX4 B-ALL B-ALL::DUX4 B-ALL::DUX4 B-ALL
## 3 S003 B-ALL::ETV6-RUNX1 B-ALL B-ALL B-ALL::ETV6-RUNX1 B-ALL
## 4 S004 B-ALL B-ALL B-ALL B-ALL B-ALL
## 5 S005 B-ALL::DUX4 B-ALL B-ALL::DUX4 B-ALL::DUX4 B-ALL
## 6 S006 B-ALL::ETV6-RUNX1 B-ALL B-ALL::DUX4 B-ALL::ETV6-RUNX1 B-ALL
## # ℹ 3 more variables: ALMA.Lineage <chr>, match.Class <chr>,
## # ALMA.match.class <chr>
Build diagnosis subset plots
ALLDataOnly <- allLeukData %>%
filter(Diagnosis %in% c("B-ALL", "T-ALL"))
p_all_only <- ggplot(
data = ALLDataOnly,
aes(
axis1 = mcf,
axis2 = Molecular.Diagnosis,
axis3 = ALMA.v0.1.14
)
) +
scale_x_discrete(
limits = c("MARLIN", "DX", "ALMA")
) +
geom_alluvium(
aes(fill = Molecular.Diagnosis),
alpha = 0.8,
width = 0.275,
discern = TRUE
) +
geom_stratum(
width = 0.275,
fill = "grey95",
color = "grey40",
discern = TRUE
) +
geom_text(
stat = "stratum",
aes(label = after_stat(ifelse(
is.na(stratum) | trimws(stratum) == "",
NA_character_,
gsub("::", "\n", stratum, fixed = TRUE)
))),
size = 2.1,
lineheight = 0.9,
min.y = 2,
na.rm = TRUE
) +
scale_fill_manual(values = metClassColors) +
labs(
title = "ALL-only alluvial",
x = NULL,
y = "Sample count"
) +
theme_minimal(base_size = 11) +
theme(
legend.position = "none",
panel.grid.major.x = element_blank()
)
AMLDataOnly <- allLeukData %>%
filter(Diagnosis == "AML")
p_aml_only <- ggplot(
data = AMLDataOnly,
aes(
axis1 = mcf,
axis2 = Molecular.Diagnosis,
axis3 = ALMA.v0.1.14
)
) +
scale_x_discrete(
limits = c("MARLIN", "DX", "ALMA")
) +
geom_alluvium(
aes(fill = Molecular.Diagnosis),
alpha = 0.8,
width = 0.275,
discern = TRUE
) +
geom_stratum(
width = 0.275,
fill = "grey95",
color = "grey40",
discern = TRUE
) +
geom_text(
stat = "stratum",
aes(label = after_stat(ifelse(
is.na(stratum) | trimws(stratum) == "",
NA_character_,
gsub("::", "\n", stratum, fixed = TRUE)
))),
size = 2.1,
lineheight = 0.9,
min.y = 2,
na.rm = TRUE
) +
scale_fill_manual(values = metClassColors) +
labs(
title = "AML-only alluvial",
x = NULL,
y = "Sample count"
) +
theme_minimal(base_size = 11) +
theme(
legend.position = "none",
panel.grid.major.x = element_blank()
)
ALL-only plot
p_all_only
## Warning in to_lodes_form(data = data, axes = axis_ind, discern =
## params$discern): Some strata appear at multiple axes.
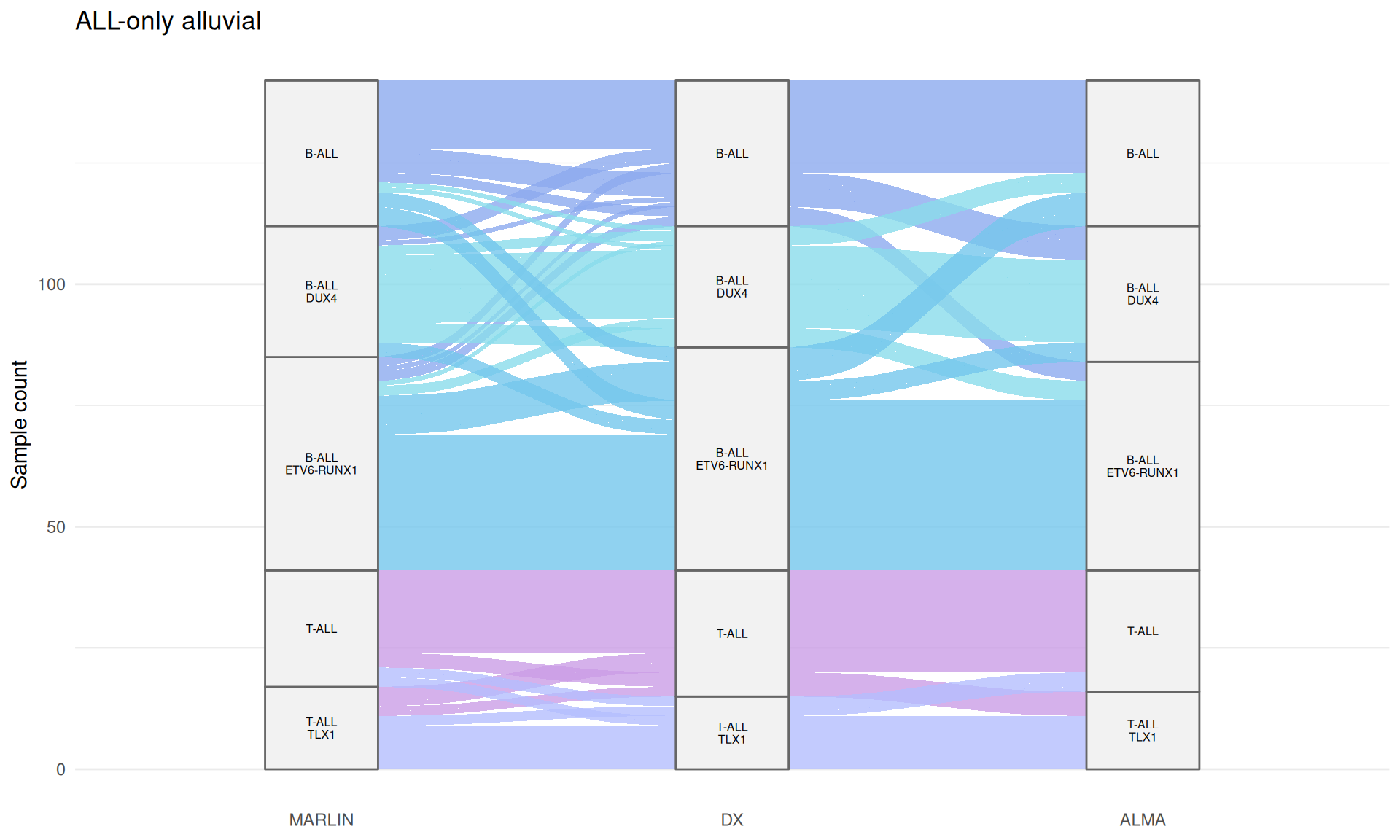
AML-only plot
p_aml_only
## Warning in to_lodes_form(data = data, axes = axis_ind, discern =
## params$discern): Some strata appear at multiple axes.
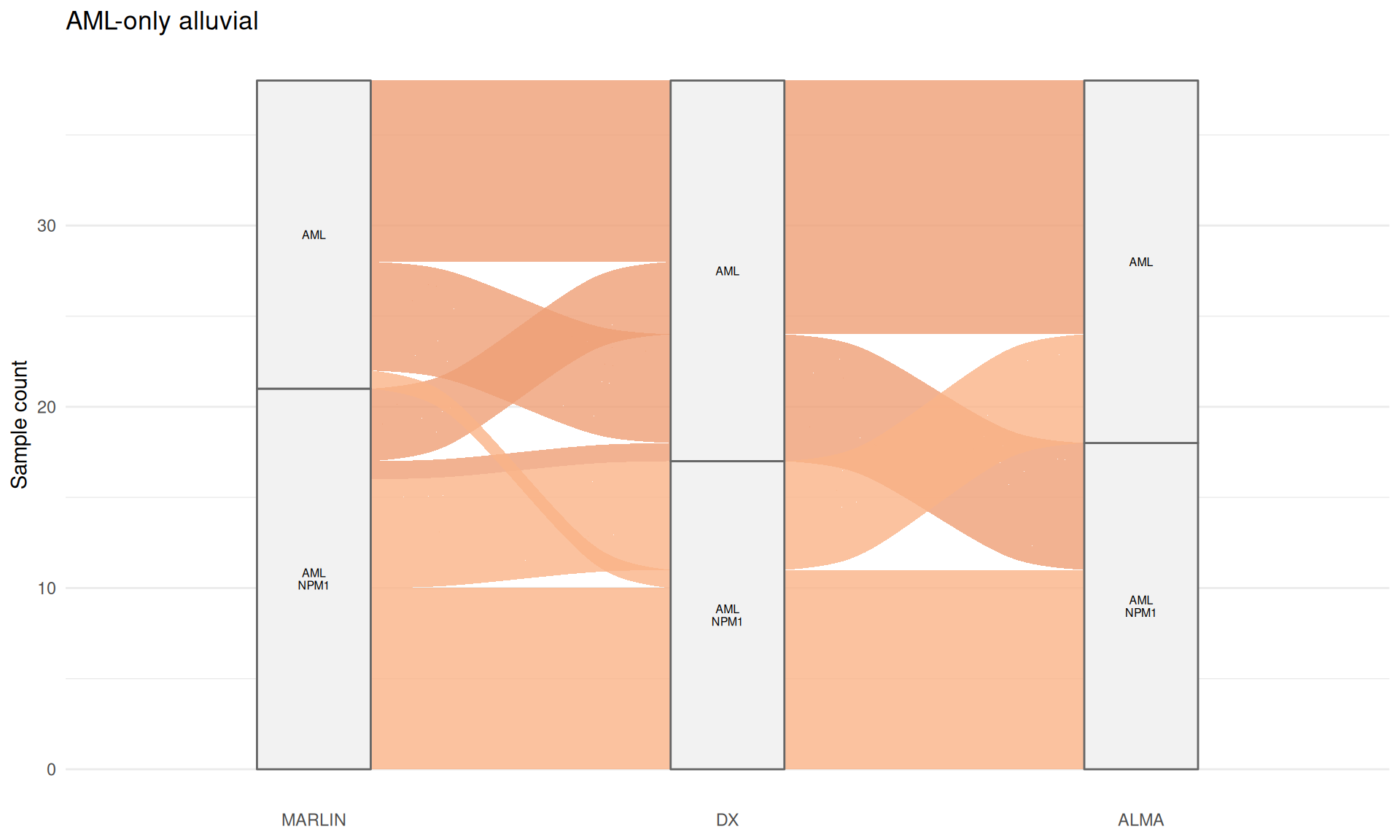
Main alluvial plot (combined)
p_main <- ggplot(
data = allLeukData,
aes(
axis1 = mcf,
axis2 = Molecular.Diagnosis,
axis3 = ALMA.v0.1.14
)
) +
scale_x_discrete(
limits = c("MARLIN", "DX", "ALMA")
) +
geom_alluvium(
aes(fill = Molecular.Diagnosis),
alpha = 0.75,
width = 0.275,
discern = TRUE
) +
geom_stratum(
width = 0.275,
fill = "grey92",
color = "grey40",
discern = TRUE
) +
geom_text(
stat = "stratum",
aes(label = after_stat(ifelse(
is.na(stratum) | trimws(stratum) == "",
NA_character_,
gsub("::", "\n", stratum, fixed = TRUE)
))),
size = 2.2,
lineheight = 0.9,
min.y = 3,
na.rm = TRUE
) +
scale_fill_manual(
values = metClassColors
) +
labs(
title = "Alluvial Comparison (Combined)",
subtitle = "MARLIN vs DX vs ALMA",
x = NULL,
y = "Sample count"
) +
theme_minimal(base_size = 12) +
theme(
legend.position = "none",
panel.grid.major.x = element_blank(),
panel.grid.minor = element_blank(),
axis.text.x = element_text(face = "bold")
)
p_main
## Warning in to_lodes_form(data = data, axes = axis_ind, discern =
## params$discern): Some strata appear at multiple axes.
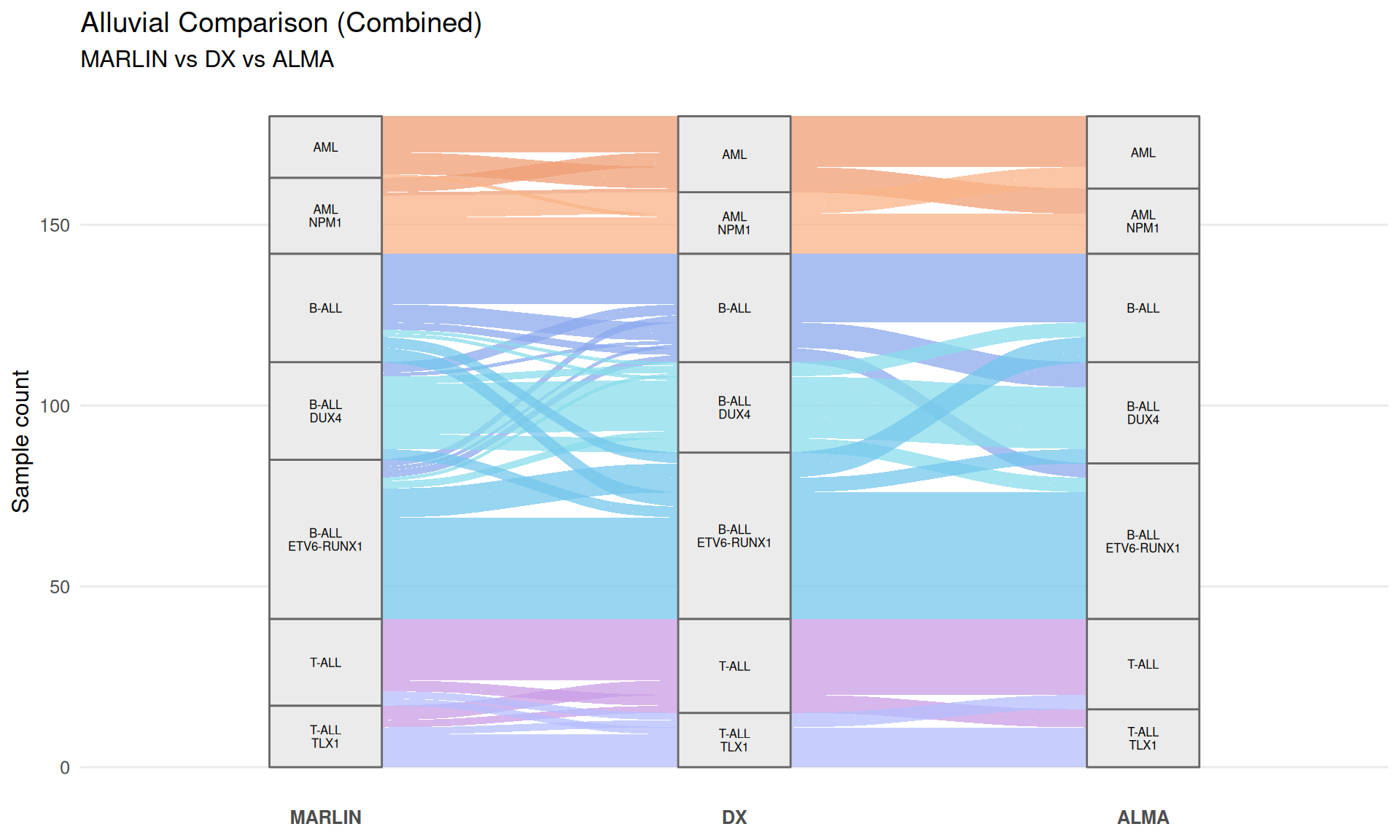
Save combined plot image
# Export the plot
ggsave(
filename = "alluvial_comparison_test.png",
plot = p_main,
width = 10,
height = 12,
dpi = 150
)
## Warning in to_lodes_form(data = data, axes = axis_ind, discern =
## params$discern): Some strata appear at multiple axes.
Session Info
## R version 4.5.2 (2025-10-31)
## Platform: x86_64-pc-linux-gnu
## Running under: Linux Mint 22.2
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.12.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.12.0 LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_CA.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_CA.UTF-8 LC_COLLATE=en_CA.UTF-8
## [5] LC_MONETARY=en_CA.UTF-8 LC_MESSAGES=en_CA.UTF-8
## [7] LC_PAPER=en_CA.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_CA.UTF-8 LC_IDENTIFICATION=C
##
## time zone: America/Toronto
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] patchwork_1.3.2 ggalluvial_0.12.6 ggplot2_4.0.2 dplyr_1.2.0
##
## loaded via a namespace (and not attached):
## [1] gtable_0.3.6 jsonlite_2.0.0 compiler_4.5.2 tidyselect_1.2.1
## [5] tidyr_1.3.2 jquerylib_0.1.4 scales_1.4.0 yaml_2.3.12
## [9] fastmap_1.2.0 R6_2.6.1 labeling_0.4.3 generics_0.1.4
## [13] knitr_1.51 tibble_3.3.1 bookdown_0.46 bslib_0.10.0
## [17] pillar_1.11.1 RColorBrewer_1.1-3 rlang_1.1.7 utf8_1.2.6
## [21] cachem_1.1.0 xfun_0.56 sass_0.4.10 S7_0.2.1
## [25] otel_0.2.0 cli_3.6.5 withr_3.0.2 magrittr_2.0.4
## [29] digest_0.6.39 grid_4.5.2 lifecycle_1.0.5 vctrs_0.7.1
## [33] evaluate_1.0.5 glue_1.8.0 farver_2.1.2 blogdown_1.23
## [37] purrr_1.2.1 rmarkdown_2.30 tools_4.5.2 pkgconfig_2.0.3
## [41] htmltools_0.5.9
